并行计算Large scale Parallel Simulator:MPI
兼容度宽:
LAMMPS is a classical molecular dynamics code with a focus on materials modeling. It's an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator.
LAMMPS has potentials for solid-state materials (metals, semiconductors) and soft matter (biomolecules, polymers) and coarse-grained or mesoscopic systems. It can be used to model atoms or, more generically, as a parallel particle simulator at the atomic, meso, or continuum scale.
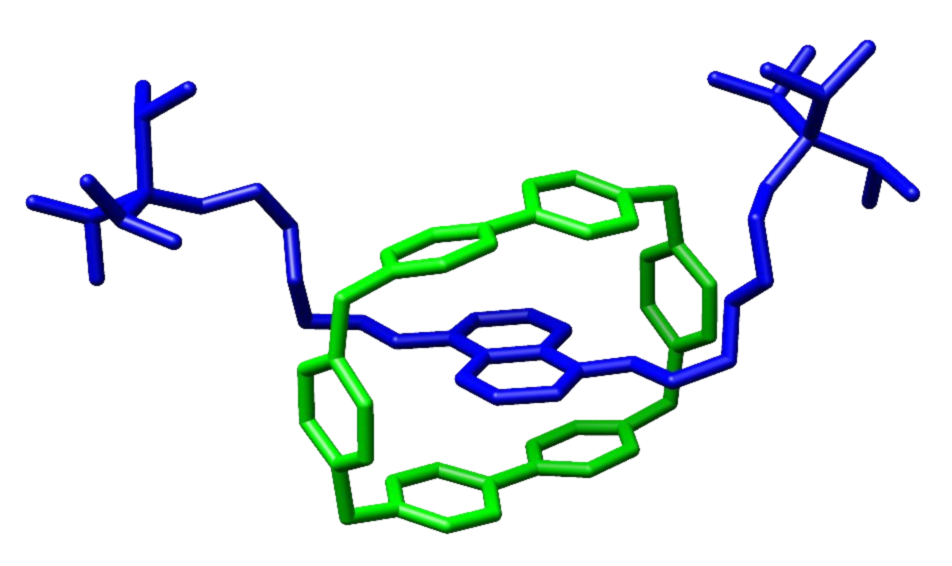
深度解读2016诺贝尔化学奖:分子马达与纳米火箭 -- 中国科学院苏州纳米技术与纳米仿生研究所
分子穿梭机【“轮烷”rotaxane】→分子开关,分子存储器,分子催化剂,分子马达,分子行走装置(如生物体内的驱动蛋白;布朗棘轮)→分子流水线 微米火箭 分子泵